
基因组编辑涉及基因组中特定 DNA 序列的删除、插入或修改。研究人员一直试图开发简单高效的基因组编辑工具以解决各类问题。
例如,如果能够简单地敲除导致某种遗传疾病的基因,基因治疗就可以迅速发展;或者,我们可以编辑作物的 DNA 来提升产量和控制病害;同样,细菌基因组也可以被修改以满足不同的工业应用。
终于,研究人员的努力取得了成功,开发出了 CRISPR 这一几乎可以编辑任何 DNA 位点的分子生物学工具。由于 CRISPR 易于使用并且功能丰富,它已经被广泛应用于各个领域,并且将在未来产生更深刻的影响与变革。
快在这里了解和学习到一切与 CRISPR 相关的基础知识,然后开启你的 CRISPR 之旅吧!
一、了解CRISPR/Cas 9 技术
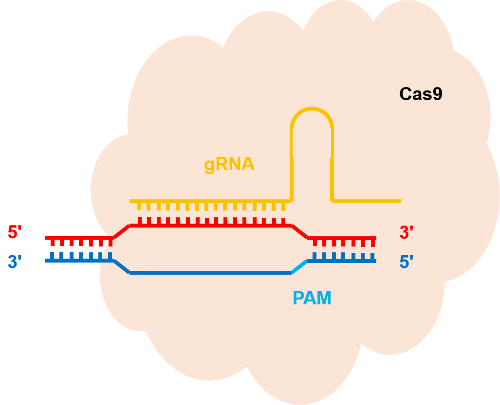
CRISPR-Cas9系统包括一个 gRNA 和 Cas9核酸酶,它们共同形成一个核糖核蛋白(RNP)复合物。 gRNA 需要基因组 DNA 中存在特定的原间隔基序(PAM)才可以结合目标序列。然后,Cas9 核酸酶在 DNA 中产生一条双链断裂(用剪刀表示)。由双链断裂所触发的 NHEJ 修复机制可能导致基因敲除。如果存在外源 DNA 模板,则可能通过 HDR 修复机制导致基因敲入。
CRISPR 系统包含两个成分: 可以识别特定 DNA 序列的 gRNA 和负责切割 DNA 序列的 CRISPR 相关核酸内切酶(Cas 蛋白)。在 CRISPR 实验中, gRNA 和 Cas 蛋白会形成核糖核蛋白(RNP)复合物。
Cas 蛋白可以被理解为是一把分子剪刀,而 gRNA 则是用于定位的 GPS 系统。在原核细胞中,gRNA 将内切酶引导至病毒 DNA。而在实验中,通过设计 gRNA 我们几乎可以定位任何有机体基因组上的任何位置。
在许多基因组工程运用中,我们使用的是源于 Streptococcus pyogenes 的 Cas9 蛋白。gRNA 与基因组目标位点的结合还同时取决于位于目标位点下游的一段前间隔序列邻近基序(PAM)的存在。其中,PAM 序列所在的 DNA 单链与 gRNA 所结合的 DNA 单链是两条不同的链。源于不同原核生物的 Cas 蛋白识别不同的 PAM 序列。我们最常用的 Cas9 蛋白识别的 PAM 序列是 5'-NGG-3',其中 N 代表任意核苷酸。
如果 PAM 序列匹配正确,并且 gRNA 成功地与目标位点结合,那么 Cas9 会在 PAM 序列上游约 3-4 个核苷酸进行 DNA 双链的切割。
非同源末端连接(NHEJ)
如果实验的目的是永久性地敲除一个基因,使其不再能够翻译得到具备正常功能蛋白,那么就可以利用非同源末端连接(NHEJ)修复机制。NHEJ 可以将 DNA 末端重新连接在一起,但这个过程中容易产生错误,并可能插入或删除核苷酸(称为INDELS)。如果插入或删除的核苷酸数不能被 3 整除,那么它将引起移码突变,使得该基因无法转录翻译得到有功能的蛋白。
同源定向修复(HDR)
如果实验的目的是将原本基因组上的一段序列替换为新的一段序列的话,那么就可以利用同源定向修复(HDR)机制。在导入 CRISPR 组分的同时还需要导入一段带有我们想要敲入的序列的外源 DNA,同时需要有对应的同源臂。细胞会利用这段外源 DNA 来同源重组,进而修复 CRISPR 系统导致的 DNA 双链断裂。这一过程中将会发生外源 DNA 序列的敲入。
二、了解 gRNA
gRNA 是一种特殊的 RNA 序列,它识别目标 DNA 区域,并将 Cas 核酸酶引导到那里进行编辑。gRNA 由两部分组成:CRISPR RNA(crRNA),一个与目标 DNA 互补配对的17-20 碱基长度的核苷酸序列;以及一个 tracr RNA,可以作为支架帮助 Cas 核酸酶折叠。
gRNA 的 crRNA 部分是可定制的组件,可以在不同的 CRISPR 实验中实现特异性。在实验中,可以将定制的 crRNA 和不变的 tracr RNA融合在一起得到只有一条链的 gRNA。在一些文献中也将这种融合的单链 gRNA 称为 sgRNA。然而,一些研究人员仍在使用了 cr RNA 和 tracr RNA 分开的 gRNA,这通常被称为两段式 gRNA 或简单地称为cr:tracrRNA。
基因敲除
当设计用于敲除实验的 gRNA 时,应该选择外显子中对于蛋白质功能至关重要的位点。出于这个原因,应该避免选择编码氨基酸太接近于蛋白质的N-末端或C-末端的序列作为目标位点。太靠近N-末端时细胞可能会在下游的另一个起始密码子开始转录,而太靠近C-末端可能错过编码正常功能蛋白质的必需部分。
基因敲入
因为基因敲入需要更多的组分,比如外源 DNA 片段来进行 HDR 修复,因此在设计上更加复杂。研究表明当切割位点与修补片段末端较远时,敲入效率会大幅降低。同时,如果是进行碱基编辑的话还会有更多的限制因素。
三、gRNA 的参数
设计 gRNA 需要考虑很多参数,并且不同的实验和细胞系也都存在差异。因此,了解基本的原理来分析具体实验至关重要。同时,现在也有一些在线的 gRNA 设计软件例如 Synthego 和 Benchling 的 CRISPR 设计工具都是很好的选择。
提升 gRNA 的靶向活性
从逻辑上讲,似乎最佳的 gRNA 应该只针对目标 DNA 序列,而不会和基因组中的任何其他地方发生互补配对,因此序列的互补性以及与目标位点的接近性应当是最重要的参数。但从上一章关于基因敲除和基因敲入的介绍中我们已经了解到具体的设计原则和对应的参数应当与你的具体实验有关。
2016年,Doench 等人分析了数千个 gRNA 的特异性,同时建立了全基因组的小鼠和人类文库,进而使用复杂的计算生物学工具建立了预测 gRNAS 靶向活性的模型。
减少 gRNA 的脱靶效应
Doench 的研究还得出了另一个有用的指标:脱靶效应得分。这一分数主要是表示 gRNA 会和非目标位点结合的可能性。
脱靶效应是 CRISPR 技术最被担忧的问题,也是最常常被大众媒体炒作的焦点。因此,任何有助于减少脱靶效应的改进对于 CRISPR 实验的成功至关重要,特别是如果要将这一技术运用于人体内。
多个 gRNA 提升编辑效率
虽然许多焦点通常都是在设计参数上的,但是还有一些其他方法可以提升 gRNA 的靶向活性并降低脱靶效应的。例如使用多个靶向同一基因的 gRNA 已经被证实是可以提升基因编辑效率的。
四、了解 PAM 序列
前间隔序列邻近基序(简称 PAM)是一个短的 DNA 序列(通常为2-6 碱基对长度)。PAM 是 Cas 核酸酶切割所必需的,通常在切割位点下游 3-4 个核苷酸。
有许多不同 Cas内切酶可以从不同的细菌中纯化,并且每种酶都能识别不同的 PAM 序列。例如,saCas9特异性地识别 PAM 序列5′-NNGRR(N)-3'。因此当找不到某一 PAM 序列时,研究人员可以选择另一种核酸酶,其相应的 PAM 序列可能存在于目标基因组中。
gRNA 中要 PAM 吗?
一般而言,当研究人员设计一个 gRNA 序列用于 CRISPR 实验时,他们不会在 gRNA 中加入 PAM 序列。以质粒作为载体为例,因为 CRISPR 系统是以 DNA 形式进入的,编码 gRNA 的区域如果有 PAM 序列的话将被翻译出的 Cas 蛋白切割。
但是最近有一种新的方法,使研究人员能够追踪细胞分化。这一机制涉及到设计带有 PAM 序列的 gRNA,以便它们能够以自身为目标进行切割。
考虑细菌中的 CRISPR 基本机制:编码 gRNA 的 DNA 序列不被 Cas 核酸酶本身切割,因为它们不包含 PAM 序列。研究人员反向利用了这一概念,开发出了归巢向导 RNA。它们与传统 gRNA 的区别只在于只需在 gRNA 中加入了 PAM 序列,因而具备了针对自身的 DNA 序列进行切割的能力,进而在分裂过程中形成多样化的条形码。
他们先前的研究已经表明,归巢向导 RNA 编辑结果比传统的 CRISPR gRNA 在累积突变中的差异更大。这种技术使研究人员能够通过追踪细胞的谱系。
五、选择合适的 Cas
Cas 核酸内切酶根据实验目的进行挑选。
SpCas9
SpCas9 是目前最广泛在 CRISPR 实验中运用的核酸酶。当 gRNA 与目的序列配对后,一旦 SpCas9 识别到 PAM序列 5’-NGG-3’,它就会在目标位点产生一个双链断裂。SpCas9 活性是两部分蛋白的共同作用:识别区域和核酸酶区域。一个负责感应互补的 DNA 序列,另一个则具有核酸酶结构来切割 DNA。
ScCas9
ScCas9是从 Streptococcus canis 中分离得到的。ScCas9 核酸酶的序列与SpCas9具有显著的相似性(89.2%序列同源性),但ScCas9 需要的 PAM 序列更加灵活——5′-NNG-3′。相对于SpCas9 的 5′-NGG-3′而言,ScCas9 能结合的位点数量约为前者的 4 倍,从而大大扩展了 CRISPR 编辑的基因组靶点。
Cas14a
Cas14a 靶向单链 DNA,不需要 PAM 序列用于激活,并且在结合靶序列时非特异性切割其他单链 DNA。Cas14a 需要与靶向的单链 DNA 高保真互补,这种特性使其成为识别与诊断遗传序列中单核苷酸多态性的主要候选核酸内切酶。
Cas13a
由于 RNA 是瞬时表达的,靶向 RNA 的核酸酶,而不是 DNA,将是永久性修饰细胞的一个极好的替代方案。Cas13a 核酸酶含有两个核糖核酸酶活性,分别催化 crRNA 的成熟和细菌中单链 RNA 的降解。研究人员还设计了 CAS13a 靶向哺乳动物细胞中的 mRNA。
Cas9 NLS, Cas9 2NLS
这一类 Cas 包含 SV40 核定位信号,可以将 CaS9 转运到细胞核中,适用于编辑真核细胞。我们推荐在 Cas9 蛋白的 C 端和 N 端都含有 NLS 信号的 Cas9 2NLS 作为这类实验的首选。
Cas9 nickases
Cas9 nickases 的两个切割结构域中的一个因突变而失活,导致仅能对一条 DNA 单链进行切割。只有当使用两个分别针对非目标链和目标链的切口酶时才能产生 DSB。这可以用来驱动 HDR 修复机制同时抑制 NHEJ 修复机制。
dCas9
dCas9 包含两个突变,使得 Cas9 的切割活性完全消失。这种 Cas 蛋白可以用于基因沉默(CRISPRi)和基因激活(CRISPRa)。
六、如何转染 CRISPR
转染方法一般分为物理转染、化学转染和病毒转染。
CRISPR 组分的物理转染
物理转染是在细胞膜上造成暂时的小孔,使得gRNA/Cas9 可以通过这些孔进入细胞。将 CRISPR 组分引入细胞的三种常用物理方法是电穿孔、核转染和微量注射。电穿孔和核转染利用了电脉冲在细胞膜上形成孔径,而微注射则利用针头将组分注射进入细胞膜或细胞核。
电穿孔和核转染的主要区别在于电穿孔通常只能让 CRISPR 组分进入细胞膜,而核转染可以让 CRISPR 组分直接进入细胞核。一般而言,用核转染将 RNP 直接转入细胞核是较理想的转染方式。
CRISPR 组分的化学转染
目前有多种化学方法可用于将分子转运到细胞中,包括磷酸钙、阳离子聚合物和阳离子氨基酸。将 CRISPR 组分引入细胞的最常用方法之一是脂质转染。
脂质转染首先涉及在 CRISPR 组分周围构建脂溶性结构,称为脂质体。然后,这些成分通过内吞作用被输送到细胞中,其中外侧会先包裹上内吞时带上的细胞膜,而后在进入细胞后脱落。CRISPR 组分然后逃逸内质体途径并通过细胞质扩散。与核转染和微量注射不同,脂质转染无法将 CRISPR 成分传递到细胞核。
CRISPR 组分的病毒转染
病毒载体可以通过一种称为转导的过程将 DNA 或 RNA 转移到细胞中。这个过程首先包括将 gRNA/Cas9 序列包装成病毒颗粒,然后将颗粒导入靶细胞。为了制造病毒颗粒,通常将含有 gRNA 或 Cas9 序列的质粒和含有病毒基因的质粒导入包装细胞系(例如 293 T细胞)。一旦病毒颗粒产生,它们就从包装细胞中纯化得到,然后被导入到目标细胞中。
有几种病毒可用于转导,包括慢病毒、腺病毒、腺相关病毒和疱疹病毒。为了稳定的转导,慢病毒,(包括 HIV)经常被使用,因为它们可以将其基因组整合到受感染细胞的基因组中。慢病毒对多种细胞系、原代细胞和干细胞都有效,并且可以在体内使用。腺相关病毒则是另一种常用于体内编辑的病毒,通常可以导致外源基因序列在细胞中持续表达。
七、如何分析实验结果
如何分析 CRISPR 编辑效率取决于引入到细胞中的突变类型。涉及同源定向修复(HDR)的新基因序列的编辑,可以通过例如限制性内切酶消化法(如果你的突变导致限制性内切酶位点的丢失或增加)或 PCR 产物的大小变化(如果你新插入的基因大到足够可以观察到变化)等方法进行评估。传统的 Sanger 测序或下一代测序(NGS)可用于检测单个核苷酸层面的变化。
现在你已经有了所有基础知识,开启你的 CRISPR 之旅吧!